For more Topic or contents you can click or refer to my another website named as pharmapathfinder.com

What is Incident :
Any unplanned or uncontrolled event in the form of non-compliance to the designed systems or procedures at any stage of testing, and storage of drug product due to system failure or equipment breakdown or manual error.
A laboratory Incident is an event in the laboratory that occurs for two primary reasons either due to analyst error or instrument error.
Responsibilities of the Personnel in case incidents:
Analyst :
Reporting of an incident to Head QC/ Designee Immediately, to initiate the incident report and to provide complete details associated with the incident to investigator, to preserve and store all the solutions, standards and reagents used in the analysis until investigation is completed and to implement Corrective Action Preventive Action proposed as per incident report.
Quality Control Head/ Designee :
To ensure that incident is reported to QA, to evaluate, investigate and conclude the incident, to conclude the incident with effective Corrective and preventive action (CAPA),to review and approve the incident investigation report and to check the effectiveness of CAPA proposed.
Analytical Quality Assurance Head/ Designee :
To register incident and assign a sequential number to each incident in log book in case of manual system, to monitor investigation,to approve root cause investigation, Corrective Actions and Preventive actions and to share incident report with CQA for review if required.
Requirements:
The analyst should be trained and aware of potential problems that could occur during the testing process.
The analyst should ensure the use of valid specification and control procedures/ method of analysis.
For the incident, where the root cause is clearly known or identified along with incident, then the investigation is not mandatory. Record the root cause for the incident in the incident form and proceed further after taking necessary corrective action. (E.g. Sequence interruption due to leak detection, vial missing, Pressure fluctuation, SST failure, data transcription error, etc.)
If the incident noticed after completion of test or observed during review and final results if come out as OOS results then the incident should be handled as per Procedure of Out of Specification.
All sample and standard solutions to be preserved as it is until completion of investigation of incident.
Laboratory investigations will be initiated within one working day of the discovery of the incident /atypical result, and will be completed within 15 working days.
Types of analyst and instrument errors (But not Limited to) :
1. Incorrect Sample
2. Incorrect Standard Preparation
3. Incorrect Sample Weighing
4. Wrong Diluent Used and Wrong Diluent Volume
5. Bracketing standard failing during chromatographic analysis
6. Mobile Phase Preparation Error or Improper Mixing of Mobile Phase
7. Improper Baseline observed, running the sample set and Improper integration observed, before release of product / Material
8. Incorrect pH Adjustment
9. Incorrect HPLC/GC Set up and Run
10. Wrong Injection Volume
11. Wrong Detector Wavelength
12. Incorrect Injection Sequence
13. Uncalibrated Instrument Used
14. HPLC/GC Auto Injector Malfunction
15. Lamp Intensity Failure
16. Column Leak, Poor Plate Count, Poor Resolution
17. Power Failure
18. Connectivity failure between software and Instrument.
19. Wrong specification selected in LIMS observed , while register the Product / material before release of product/ Material.
20. Chromatographic peaks are not eluted in between the sample set .
21. Improper separation (merging the components) observed in chromatograms, during the chromatographic analysis.
22. System failure observed during the analysis due to column high pressure/ Instrument.
23. In case of calculation errors or wrong reporting observed after release of materials/products, which effect the end value.
24. In case of bracketing of standards in assay, content uniformity, dissolution, dissolution profile any deviation from specified limits.
25. Calibration failure during quarterly/daily calibration procedure
Incident Procedure :
When an analyst obtains atypical results, or when the analyst becomes aware of laboratory errors that were committed during testing, he or she will annotate the atypical results or known laboratory error in the appropriate analytical protocol and notify section in-charge immediately.
Analyst shall preserve all starting materials, solvents, intermediate and final solutions, and standards used in the testing. These materials will be retained and preserved until completion of the investigation or until it has been determined that they are no longer useful.
The analyst shall fill all details into incident report and based on review of incident, section in-charge shall intimate to QA for login of incident report.
Based on the request of section in-charge, QA person shall enter the details of incident in laboratory Incident Register, assign a sequential number to the incident report in case of manual system.
The QC Head/Designee will review the results, the analytical protocol, the starting materials and all solutions, the raw data, and the calculations used to generate the final results.
The checklist will be completed by the QC Head/Designee and analyst together. The QC Head/Designee will interview the analyst, and will check relevant documents, in order to answer questions which will help identify whether common or obvious laboratory errors were the root cause of the Incident/atypical result.
If an assignable cause has been found, and the original result is invalidated and the analyst repeats the test. The investigation findings are documented in the incident Report and the investigation is closed.
If no assignable cause is found, a full investigation will be completed involving both manufacturing as well as the QC laboratory, since the origin of the incident /atypical result is unknown. These will be handled as full investigations since those types of results may be attributed to product quality and a full investigation is needed to determine if the result is valid or not.
If the original test solution is suspected, re-dilution of the stock or intermediate sample preparations can be re-analyzed.
a. If all re-analyzed, re-diluted results do not confirm the original result, and meets acceptance criteria, then dilution error may be the assignable cause. If this is found to be the case, then the re-dilution results from the re-analysis are accepted. The original result is invalidated and the investigation is closed.
b. If the re-dilution results do not confirm the atypical results, other factors such as insufficient mixing, sonication, shaking, etc., may be suspected as the assignable cause. The results are recorded in the investigation. The original atypical result is invalidated. Re-test the sample according to the method .
c. If any of the re-dilution results confirm the original atypical result, a full investigation is required to determine if the original results are valid or not.
If new results from the investigation are within acceptance criteria, and assignable cause is determined, the new results are accepted and reported, and the Laboratory Investigation is concluded.
When an assignable cause is found for a laboratory error, an appropriate corrective and preventative action plan (CAPA) must be taken.
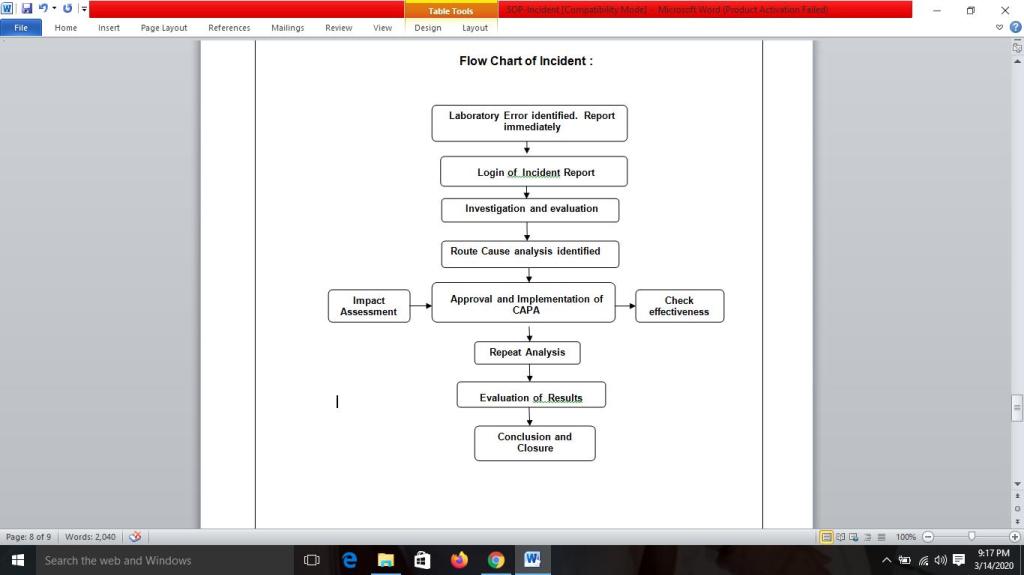
Flow chart of Incident :
Incident Check list :



CORRECTIVE ACTION AND PREVENTIVE ACTION REQUIRED FOR ABOVE Types of analyst and instrument errors
LikeLike
Yes CAPA is required for Analyst error and instruments errors
LikeLike
Good post. I learn something totally new and challenging on websites I stumbleupon every day. It’s always interesting to read through content from other authors and use something from their sites.
LikeLike
Why do we need to have SOP for handling laboratory incidence? Cant this be covered directly in deviation itself as the incident that has occurred is already deviating the set SOP rules? Any comment on this?
LikeLike
Good question dear and in some company the word Deviation and incidence are not there and such types of things are considered as Process Non Conformance (PNC) but overall the things are same but in deviation SOP we can not club all the details like Analyst error and instrument errors and in which cases we have to take incidence etc. so for that reason separate SOP for incidence along with form is better for compliance and to understand the analyst working in Quality control laboratory. Hope it will be clear for you
LikeLike
Categorization is required to investigate incident i.e Critical, Major & Minor. If yes please provide guideline if any available.
LikeLike