The FMD (Falsified Medicines Directive) is a legal framework introduced by the European Commission to improve the protection of Public health within the European Union. The directive applies since 2 January 2013 & the European Commission Delegated Regulation, (EU) 2016/161, supplements Directive 2001/83/EC with rules regarding safety features for the packaging of medicinal products for human use. The regulation was adopted in October 2015 to counteract to fake medicines include stricter record-keeping of wholesale distributors, pharmaceutical producers, an EU-wide quality mark to identify online pharmacies and mandatory safety features on packages.
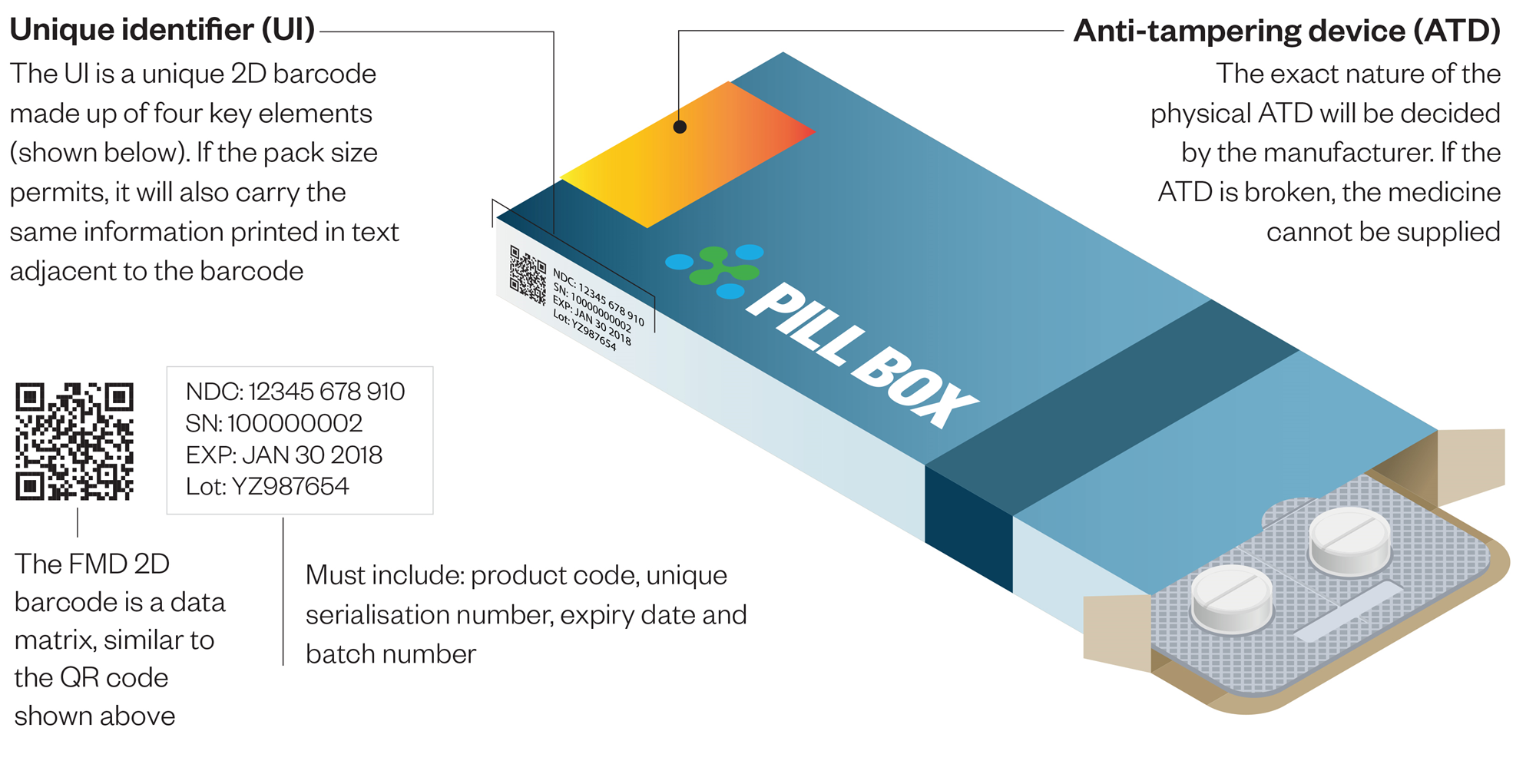
It requires that a unique identifier must be encoded in a two-dimensional barcode printed on each unit of sale package which is to contain:
- Product code
- Randomized serial number
- Expiration date
- Batch or lot number
- National Health Reimbursement Number if required
There must be a Tamper Evident Device.
When the medicine or vaccine is dispensed it must be scanned and the barcode decommissioned, so that it cannot be reused on a falsified medicine.
2,291 pharmaceutical companies with marketing authorizations to supply prescription medicines to the European Economic area are required to connect to the EU Hub established by the European medicines verification Organization and upload the unique identifier for each pack of medicine they manufacture or repackage before February 2019. By August 2018 only 841 companies had completed the first stage of connection, which may take up to six months.
Around 1,500 pharmaceutical companies are at risk of failure to comply with the Falsified Medicines Directive (FMD) because they have yet to start working with the European Medicines Verification Organization (EMVO), the body has warned.
The FMD and its track and trace model :
The FMD introduces stringent regulations aimed to improve public health with new harmonized, pan-European measures that control and monitor the trade pathway of medicines to safeguard them for human use. The FMD track and trace regulations include multiple, diverse rules for all stages of the product, distribution and dispensation lifecycle, which derive from a strong foundation of safety feature regulations and rigorous verifications.
Fundamentally, the FMD enables a point of dispense verification model for medicines. To enable this model, the law requires the implementation of several different track and trace capabilities. There are three core regulations
1: Serialization :
The FMD requires serialization at the saleable pack or secondary level. For each pack of drug product, a unique serial number, coupled with the manufacturer product code, batch number and expiration date, are to be encoded in both a GS1 2D datamatrix and in human-readable form. A fifth data element, such as a national reimbursement number, may be required based on country requirements.
2: Compliance reporting :
The Marketing Authorization Holder (MAH) has several reporting and notification requirements under the FMD. The primary regulations apply to product master data and serialized product pack data. First, master data about the product, including product codes, form, strength, doses per pack, pack type, and target market(s) for distribution, must be reported to the European hub for each unique product form produced along with any subsequent updates. Serialized product pack data must also be reported, including product codes, lot/batch number, expiry data and serial numbers, for each unit of drug product shipped into the supply chain. Drug product status must also be maintained and updates made to the EU hub. These status updates may be required at a batch level if a recall is initiated, or at a saleable unit level in situations such as the decommissioning of serial numbers due to destruction of drug product.
3: Verification and safety features :
The FMD requires verification of the safety features, including the serialized product identifier, at least once before the product leaves the supply chain and is dispensed to the patient. The drug package barcode is scanned, and the information is submitted in a query to a national repository system which contains data about the drug package that was originally submitted by the manufacturer or marketing authorization holder. The national repository checks to verify that the product code and serial number of the product scanned matches an active unique identifier in the system. Under simple distribution when the product moves from MAH to wholesaler to pharmacy, this verification is performed by the pharmacy dispenser.
Simple in concept, complex in implementation :
The FMD creates an umbrella harmonized regulation covering the member states of the EU, plus four other countries (Norway, Iceland, Liechtenstein and Switzerland). The law also provides flexibility in how the regulations apply for drug products targeted for dispensation within a given country. So, a pharmaceutical company preparing for FMD regulations needs to design their serialization and compliance infrastructure both for the extreme scalability challenges presented by the FMD and the flexibility required to serve the member states. For example, a drug product may be regulated as a prescription medicine in one member state but not another, thereby creating serialization requirements for some drug packages and not others.
Certain prescription drug products may be white-listed, or exempted, from the safety feature requirements. A member state may follow the standard GS1 GTIN to identify the drug product, or they may require a unique national product code to be used. Member states may also require additional data to be captured, stored and reported with each drug product, such as a national reimbursement number. These are some of the complexities facing a pharmaceutical company preparing their internal packaging sites and external CMO network, and the CMO looking to serve a diverse pharma client base when preparing for FMD compliance.
Managing the network becoming bigger challenges :
Implementing a secure compliance infrastructure for EU FMD means that pharmaceutical companies and their supply network partners are challenged with mastering a never-before-seen level of network management .
European citizens are entitled to medicines that are safe, of high quality and effective. Falsified medicines may contain ingredients of low quality or in the wrong dosage – either too high or too low – and therefore pose a major health threat to EU citizens. The Falsified medicines Directive– which entered into force on 2 January 2013 – makes medicines safer by providing measures to verify their authenticity and improve the quality of their ingredients. The main novelties are three:
- First, prescription medicines will need to bear, on their outer packaging, a pack-specific number and an anti-tampering device that will allow the pharmacist to verify that the medicine is authentic and unopened before dispensing it. This will prevent falsified medicines from reaching the patients.
- Second, the active ingredients of medicines are to be manufactured according to appropriate quality standards (“good manufacturing practice for active substances”) regardless of whether they are manufactured in the EU or imported. If imported, the country of origin has to certify that the active ingredient has been manufactured according to standards equivalent to those of the Union. These provisions ensure that only safe, high quality ingredients are used in medicines in the EU.
- Third, legitimate online pharmacies will be identified by the same logo across the EU. The logo, when clicked, will allow the verification of the legitimacy of the pharmacy. This will allow EU citizens to make an informed choice when buying medicines over the internet.
The Falsified Medicines directive marks a real breakthrough for the safety and quality of medicines circulating in the EU, not only it will be more difficult for falsified medicines to reach patients, but also European citizens will be able to buy medicines online through verified sources. Furthermore the Directive will also ensure the use of only high quality of ingredients in the composition medicines that have a beneficial effect on the level of public health protection in the EU.
Fake medicines entering the legitimate medicines supply chain is a growing problem. Such occurrences are difficult to detect but, according the Medicines and Healthcare products Regulatory Agency, since 2004 there have been nine known cases of fake prescription-only medicines reaching patients through the legal supply chain in the UK.
To tackle this problem the EU adopted Directive 2011/62/EU, known as the “Falsified Medicines Directive”, in July 2011. The Directive aims to prevent falsified medicines entering the supply chain and reaching patients by strengthening all aspects of the manufacture and supply chain across Europe. It introduces increased regulatory requirements for suppliers, manufacturers and wholesalers.
What is a unique identifier?
Medicines packaging will have to contain a unique identifier so that prescription medicines can be verified, and individual packs can be tracked through the supply chain. The European Commission is currently consulting on a concept paper about the technical specifications of this unique identifier, and is considering three options: a traditional linear barcode, a two-dimensional (2D) matrix barcode and radiofrequency identification (RFID). A limitation of linear barcodes is that they are unable to carry large amounts of information. 2D matrix barcodes can hold much more data but, currently, many pharmacies in Europe including in the UK are not equipped with the scanners required to read them.
An RFID (Radio-Frequency Identification) An RFID (Radio-Frequency Identification) are a type of tracking system that uses smart barcodes in order to identify items. These radio waves transmit data from the tag to a reader, which then transmits the information to an RFID computer program.
How will this work?
It is proposed that the serial numbers (contained in the unique identifiers) for all medicines will be checked in to a data repository by manufacturers and then, at the point of dispensing, a pharmacist will scan each unique identifier and check out the medicine being dispensed.
What will need to be authenticated?
Not all medicines will require a unique identifier. The Directive states that all prescription medicines will need to be authenticated, unless they are included in a “white list” of medicines that are deemed not to be at risk of falsification. Conversely, medicines that do not require a prescription will not need a unique identifier unless they are put on a “black list”, in which case they will need to be authenticated before supply. Details about how the black and white lists will be compiled are part of the consultation process and, consequently, are not yet known.
How FMD Works & its Features :

How medicines verification will work in practice :
Manufacturers will give all their packs unique serial numbers and packs will carry a code that includes what the product is, the serial number, batch and expiry data. Depending on the market, there may also be a reimbursement number, although this is not relevant in the UK. This will all be encoded in a 2D barcode or matrix printed on each pack.
Manufacturers will upload this pack data to the European Medicines Verification System (EMVS), a central information hub and data router. As medicines are shipped to their point of sale, such as the UK, data from the EMVS will be transferred to the UK’s National Medicines Verification System (NMVS).
The FMD applies to prescription-only medicines as well as some over-the-counter medicines where there is a track record of counterfeiting. The package will be tracked from manufacturer through to the pharmacy, where it will be dispensed to the patient.
The pharmacist will scan the code on the pack and send the data to the NMVS (National Medicines Verification System) & In the vast majority of cases, this should return a message saying that the code matches one uploaded by a manufacturer and the pack is genuine. The pharmacist will then decommission the pack changing its status in the system to ‘inactive’ before handing the medicine to the patient.
“However, if the message comes back negative, there should be a reason for this, such as [the pack] has been recalled, withdrawn or dispensed elsewhere, and the pharmacist will have to take appropriate action.
Advantages of FMD :
Once the NMVS is in place or at least once some technical specifications are made available pharmacy software system suppliers will need to build their interfaces to the national system. Pharmacies will need to invest in scanners and start to understand how workflows will change. New standard operating procedures will need to be written. There will be training requirements, as well as a need to understand how compliance with the FMD will be regulated.
Disadvantages of FMD :
This is a system originating from Europe that may not take account of UK anomalies. In Europe, for example, medicine packs are generally not split. But in the UK, packs are often opened and strips of tablets are shared between patients. Such practice does not sit easily with a system where a pack must be scanned and dispensed to a patient if a box has been scanned once and the code decommissioned, a second scan for a second patient will trigger an alert.
For similar reasons, it is not clear how the FMD will work when community pharmacists prepare a seven-day supply of multiple medications for patients with co-morbidities.
Conclusion :
Timetable is not as tight as it looks. The FMD applies only to medicines manufactured after the 9 February 2019 implementation date and these won’t be in hospital or community pharmacy for quite some time & it will take years to get the system up and running.
There is a long way to go before anyone has a clear idea of what FMD in practice will look and feel like. There’s a mountain of work to do and a deadline by which to do it. The only certainty is this pharmacy processes are about to change again.

ok
LikeLike
ok
LikeLike